
近日,香港科技大学黄湧教授与深圳湾实验室陈杰安研究员团队在手性合成与药物分子砌块领域取得重要突破,相关研究成果以Harnessing thia-Rh-carbenes for the enantioselective synthesis of chiral α,α-diheteroatomic carboxylic acids为题,在线发表于国际顶级催化期刊《自然 催化》(Nature Catalysis)。
在化学、化学生物学与创新药研发领域,精准调控生物活性分子的结构与功能,是贯穿始终的核心目标。手性α-杂原子羧酸是调控肽、蛋白质及其他生物活性分子结构与功能的核心单元,其中最广为人知的便是构成生命基础的α-氨基酸——其核心特征便是α-碳原子上连接1个氮杂原子,是蛋白质、多肽药物的核心“分子积木”。而其结构与功能的“升级版”——α,α-双杂原子羧酸,即在同一个α-碳原子上连接两个不同的杂原子(氧、氮、硫等),则展现出更为巨大的应用潜力。这类分子凭借独特的电子特性与非共价相互作用,能在不显著增加分子质量的前提下,精准调控分子的二级结构与理化性质,是优化药物构象、提升生物活性、规避耐药性的理想砌块。但数十年来,这类分子的合成与应用始终处于空白状态,核心瓶颈集中在两大难题:1.中间体可控生成难:合成所需的杂原子取代金属卡宾中间体极不稳定,难以实现精准、可控的原位生成,传统方法仅能实现单杂原子的引入,无法完成同一碳原子上双杂原子的精准连接;2.手性精准控制难:双杂原子带来的高极性、杂原子密集的化学环境,会引发复杂的氢键、偶极作用与副反应,导致手性无法精准调控。
针对上述挑战,研究团队另辟蹊径,开发了一套兼具原创性与通用性的合成新策略,从根源上破解了α,α-双杂原子羧酸的合成瓶颈。
1. 设计α-硫代铑卡宾合成体系,实现高选择性双杂原子引入
团队设计并合成了稳定易制备的S,S-双叶立德底物,该底物与桨轮状Rh(II)催化剂反应后,可在温和条件下原位生成此前难以获得的α-硫代铑卡宾中间体,解决了杂原子取代金属卡宾中间体不稳定的核心挑战。在此基础上,团队筛选出两类专属手性质子梭催化剂:针对 O-H键插入反应,筛选出Takemoto手性氨基硫脲衍生物CAT-1;针对N-H键插入反应,筛选出螺环手性磷酸CPA-9。两类催化剂解决高极性环境下的手性控制难题,成功实现了高对映选择性的O-H、N-H键插入反应,以优异的收率和极高的手性纯度(e.e.值最高可达98%),成功构建了α,α-O,S、α,α-N,S型双杂原子羧酸酯产物。该反应体系条件温和(常温/40 ℃、中性环境),底物兼容性极强,兼容芳基、烷基、复杂天然产物片段等数十种底物结构,官能团耐受性优异,且可实现克级放大生产,放大后产物的收率与手性纯度无明显损失,具备突出的工业化应用潜力。
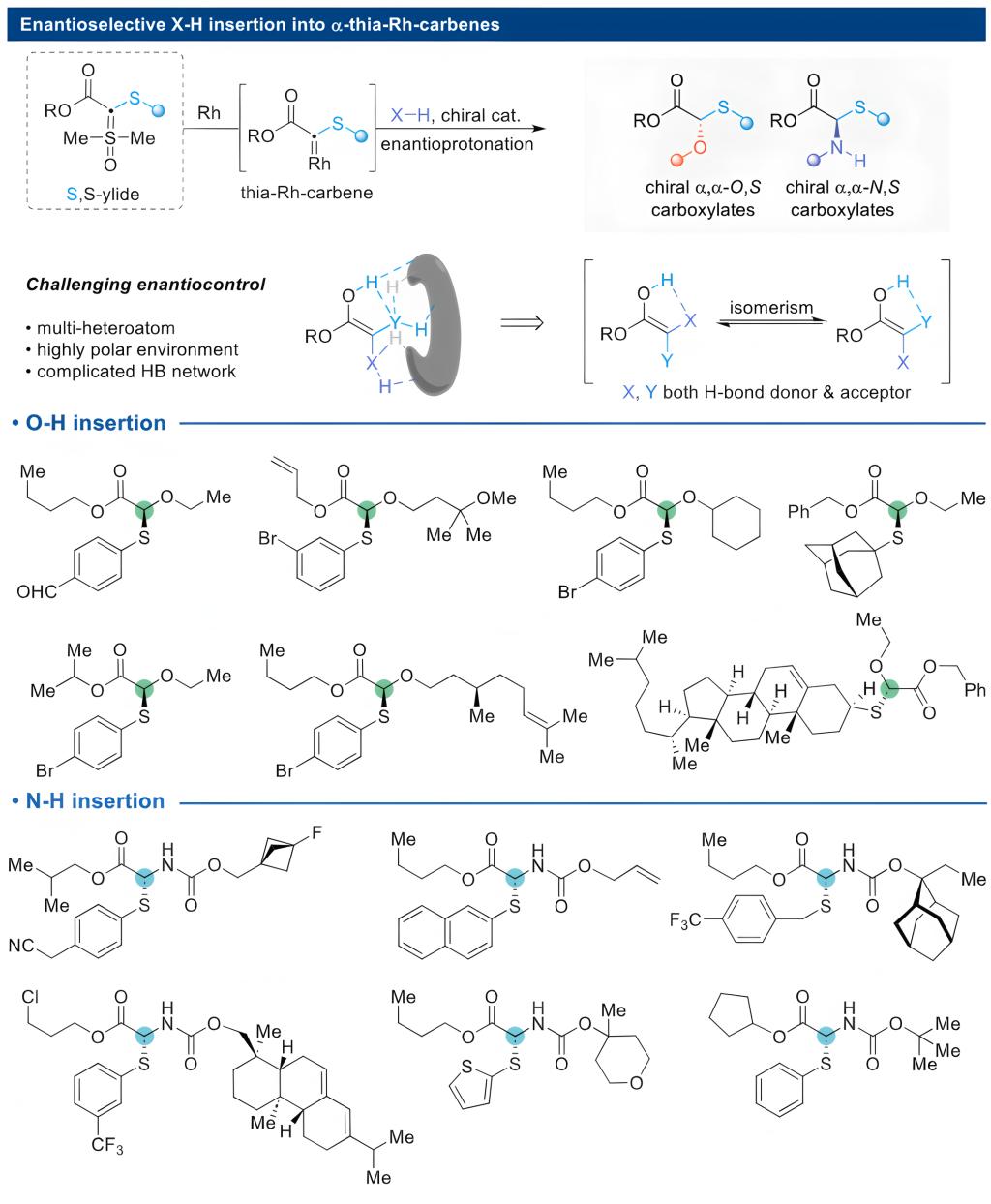
随后,团队对反应手性决定步骤“不对称质子化”进行了密度泛函理论(DFT)计算研究。反应优先通过低能垒的[1,3]-质子转移,生成动力学与热力学均更稳定的Z-烯醇中间体。手性磷酸可通过非共价相互作用,与Z-烯醇中间体形成紧密的作用网络,精准诱导其发生对映选择性质子化。基于AIM理论的电子密度拓扑分析,对过渡态的电子密度进行了定量计算。结果显示:生成优势R-型产物的过渡态TSZ-R存在多达9处关键非共价相互作用(包含独特的N-H···π相互作用),而竞争路径的过渡态仅存在5处弱相互作用。前者在能量上比后者低3.1 kcal/mol,从理论层面解释了反应实现高对映选择性的核心原因。
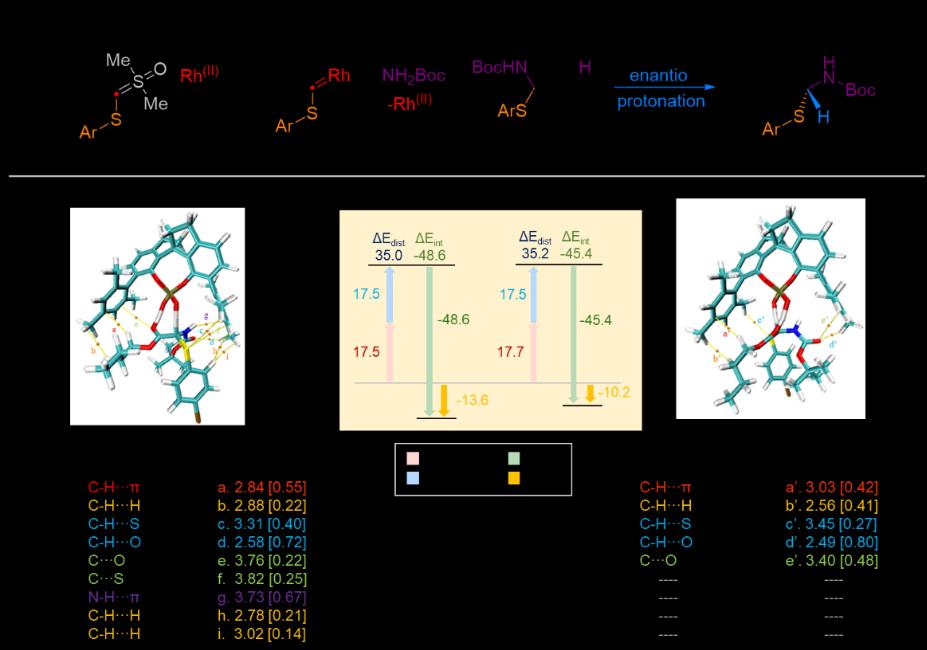
在核心合成体系的基础上,团队进一步开发了“硫原子交换”通用策略,能够将产物中的α-硫原子立体选择性地替换为氧、氮等其他杂原子,从而快速衍生出结构多样的α-杂原子非天然氨基酸库(α-Het UAA)。所获得的α-oxy和α-amino UAA是一类此前难以获得的非天然氨基酸。这一转化反应兼容多种N-保护基修饰,同时保持高对映选择性,并适用于源自复杂药物分子的醇类化合物,展现出广泛的官能团耐受性。同时,α,α-双杂原子羧酸酯可作为多功能的手性结构单元,轻松转化为多种官能团,例如羧酸、Weinreb酰胺、酰胺、酮、炔酮和醇,这些化合物均源自同一前体。
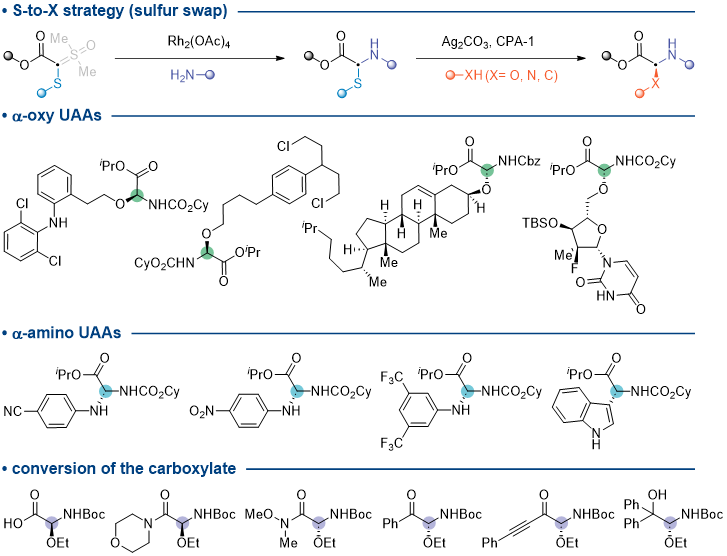
为了验证合成产物的结构独特性与应用潜力,团队通过数据驱动的方法,将合成的31种α-杂原子非天然氨基酸,与天然氨基酸、314种多肽合成常用的常规非天然氨基酸进行了系统的化学空间对比。t-SNE降维可视化分析结果清晰显示,本研究合成的α-杂原子非天然氨基酸,在化学空间中形成了与天然氨基酸、常规非天然氨基酸完全区隔的“独特性孤岛”,扩展了现有非天然氨基酸的化学版图。同时,主成分分析(PCA)进一步证实:尽管这类分子结构新颖、带有双杂原子特征,但其理化性质与天然氨基酸保持了高度的相似性与生物相容性。这一特性意味着,这类分子可无缝接入现有的多肽合成、蛋白质修饰体系,无需对现有工艺进行大幅改造,具备极强的应用落地潜力。
本研究的核心价值,不仅在于学术层面的方法学突破,更在于其在多肽和药物修饰化学的应用潜力。所合成的新型非天然氨基酸可高效实现与天然氨基酸的无消旋肽键偶联,兼容Phe、Met、Trp、Pro等绝大多数天然氨基酸,可精准用于多肽的定点修饰与功能调控;同时可对舍曲林、西格列汀、缬苯那嗪等多款上市药物进行后期结构修饰,在中枢神经疾病、代谢性疾病药物优化中同样具备广阔的应用前景。

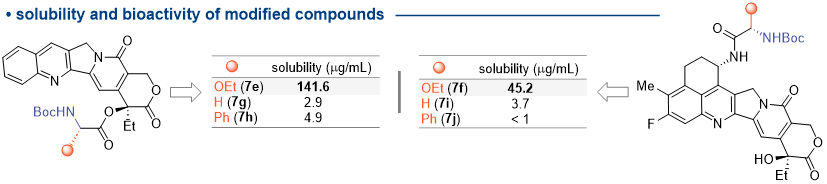
特别值得关注的是,α-杂原子非天然氨基酸的引入在解决抗癌药行业两大核心痛点——水溶性差和肿瘤耐药性方面展现出了良好的效果。喜树碱和依喜替康是临床广泛应用的经典拓扑异构酶抑制剂,也是抗体偶联药物(ADC)中最常用的细胞毒素载荷。然而,它们的强疏水性导致水溶性极差、体内生物利用度低,并易产生耐药性,这些问题一直是临床应用的核心瓶颈。团队利用本研究合成的手性α-乙氧基非天然氨基酸模块,对喜树碱和依喜替康进行了精准的结构修饰,从而实现了理化性质的微调:
水溶性实现数十倍跃升:修饰后的药物类似物7e、7f在水中的溶解度分别达到141.6 μg/ml、45.2 μg/ml,相较于对应的甘氨酸修饰衍生物(2.9 μg/ml)、苯丙氨酸修饰衍生物(<1 μg/ml),溶解度分别提升了49倍、45倍以上,解决原药水溶性差的痛点;抗肿瘤活性与耐药规避能力大幅提升:体外细胞实验显示,7e、7f在8种人类癌细胞株中,半数抑制浓度(IC50)普遍降至200 nM以下,展现出强效广谱的抗肿瘤活性;在DU-145、HepG2等6种耐药、转移性癌细胞株中,其抑制活性比传统氨基酸修饰衍生物高出5-10倍,在依喜替康耐药的DLD-1细胞株中依然保持强效活性,突破传统药物的耐药机制。
尤为重要的是,这一成果直接挑战了“药物活性优化主要依赖提升分子亲脂性”的传统药物设计理念,证实了通过α位双杂原子引入极性,反而能同时提升药物的水溶性与靶向活性,为创新药分子设计提供了全新的思路。
总的来说,该研究首次建立了α-硫代铑卡宾介导的不对称X-H插入反应体系,成功合成了以往无法获得的手性α,α-双杂原子羧酸(包括α,α-O,S、α,α-N,S、α,α-O,N、α,α-N,N四大类),从根本上解决了同一碳原子上精准引入两个不同杂原子的难题。该研究拓展了金属卡宾不对称插入反应的应用边界,首次实现了单碳原子上双杂原子的可编程、不对称引入,丰富了手性羧酸与非天然氨基酸的化学空间,为杂原子卡宾化学的发展提供了全新的理论与实验支撑。此外,高效的新型非天然氨基酸合成平台,为多肽药物研发、ADC药物载荷优化、蛋白质功能调控提供了全新的分子工具,有望推动多肽化学、化学生物学、创新药研发等多个领域的技术革新。
本文由香港科技大学的黄湧教授和深圳湾实验室的陈杰安研究员共同指导。香港科技大学的博士毕业生邢亚洁担任第一作者,深圳湾实验室-香港科技大学联培博士生方玉琦负责完成DFT计算部分。此外,香港科技大学的博士生赵旖澄也为本文做出了重要贡献。
原文信息:
Harnessing thia-Rh-carbenes for the enantioselective synthesis of chiral α,α-diheteroatomic carboxylic acids
黄湧课题组主页:http://www.huangresearch.org/
陈杰安课题组主页:https://chen.szptmc.cn/
撰稿 | 邢亚洁 方玉琦
编辑 | 鲍 啦
责编 | 远 山
欢迎投稿、建议 | media@szbl.ac.cn



 粤公网安备44031102000926号
粤公网安备44031102000926号